The long-term fitness consequences of early environmental conditions
May 9, 2017 Leave a comment


Adam Hayward
The conditions which an individual organism experiences during early development have a profound effect on their success in life. For example, poor maternal nutrition may lead to underdeveloped or small offspring, which are likely to have reduced fitness in later life. A host of studies in the lab and field have shown that the quality of environmental conditions (nutrition, population density, climate, predation, infection) during early life are strongly associated with body mass, survival, ageing rates, reproductive success, disease resistance and lifespan.
There are two (non-mutually exclusive) explanations for why early-life conditions influence later performance. First, a non-adaptive explanation: if conditions are good, development is good and the individual is well set-up for a successful life; if conditions are bad, development is sub-optimal in some way and the individual struggles. This is known as the ‘silver spoon’ hypothesis (although The Who would call it the ‘plastic spoon’ hypothesis), and under this scenario, a bad start in life always leads to poor performance in adulthood. Second, an adaptive explanation: the individual senses its environment during development, assumes that it reflects the environment it will encounter later on, and develops in such a way as to maximise its performance under those conditions. Under this scenario, if conditions match during early and later life, no matter how bad those conditions are, individuals will have higher fitness. This is the ‘predictive adaptive response’ hypothesis, and it has been a popular (but controversial) explanation for the origins of metabolic disease in humans.
Few studies have tested for predictive adaptive responses in long-lived wild animal populations, because it’s difficult: such a test requires measurement of environmental conditions in early life, plus measures of both environmental conditions and performance in later life. Gabriel Pigeon and colleagues, from the University of Sherbrooke in Canada, used more than 40 years of data on a bighorn sheep population to test for predictive adaptive responses in a recent paper. They concentrated on female sheep, and asked whether (1) probability of weaning a lamb and (2) probability of survival in a given year were dependent on early-life environmental conditions, current conditions, and an interaction between the two. They tested 12 different environment variables, including population density and a large number of climatic variables (although only density was important, with the hypothesis being that higher density = more competition = poorer nutrition).
They ran a large number of models for both response variables, including linear and non-linear effects of early-life variables and crucially, interactions between early-life and current conditions. They also attempted to separate out within- and between-cohort and –individual effects, which was rather cool, using a really nice approach developed a few years ago. In short, it was pretty thorough.
Population density at birth explained 32% of variation in weaning success: females born in high density years were less able to wean lambs. There was also an interaction with current population density: in high-density years, females were less likely to wean lambs, but this was only true of females who experienced high density around birth themselves. In other words, experiencing poor conditions in early life made individuals less able to deal with poor conditions in early life (Figure a below). However, population density at birth was very weakly (and non-significantly) associated with survival (Figure b below).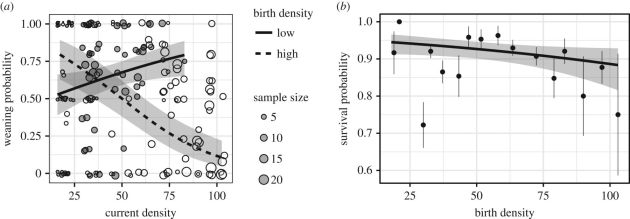
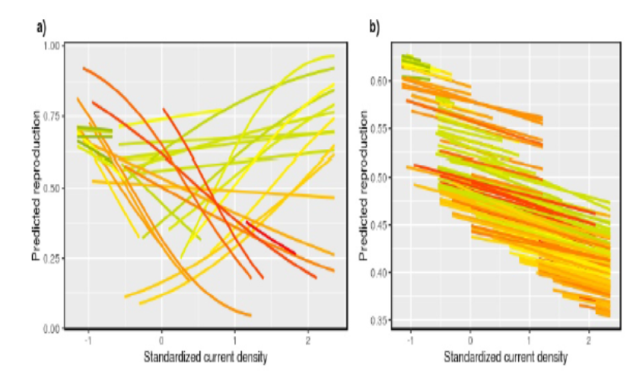
There were some interesting (if rather mind-bending) results concerning how the current population density influenced weaning success, illustrated below (in the SI, where I went digging, so you/other PEGE members don’t have to!). In (a), each line represents the change in weaning success with increasing current density in a given cohort, and the redder the line, the higher the early density was in that cohort. There are between-cohort effects, because the cohorts are responding differently to current density; however, there is no average within-cohort effect, because the average cohort would show a line with a slope of approximately zero. In (b) each line represents an individual. There are between-individual effects, because individuals who experienced higher current density had lower fitness, but there are no within-individual effects, because all individuals responded in a similar manner to increases in density. This suggests the absence of individual plasticity, and that density affects all members of a cohort in a similar way.
The main conclusion of the paper is that analyses did not support a predictive adaptive response. This is perhaps not surprising, given similar conclusions in a recent(ish) meta-analysis of experimental studies in plants and (short-lived) animals and even some not-especially-convincing (OK, it’s mine) stuff on humans using data on climate and famines. Predictive adaptive responses are an incredibly intuitive and lovely hypothesis at first glance. I’ve found this when teaching undergraduates: given the question ‘how can low nutrition during gestation lead to diabetes in later life?’, one or two will always come up with the idea of predicting the future environment. Evidence in support of such responses are rare though. An interesting question to ask is ‘how predictive is predictive?’ Do the fitness benefits of developmental plasticity need to arise when you’re halfway through life? Reaching sexual maturity? Surviving to weaning/fledging? Surviving the pre-natal period? All have been suggested, but the best examples of predictive adaptive responses (for me) are very short-term responses: one in voles, and one in humans. Are they predictive or even adaptive? It’s up for debate.